
Please forgive the length of it.
Epigenetics and Cancer
Introduction
Epigenetics is the field of study concerned with the heritable differences in gene expression without changes in the DNA sequence. Primarily this concerns the methylation of CpG dinucleotides at the 5’ C position of cytosine nucleotides, resulting in 5-methyl cytosine, and the acetylation/ deactylation of histone proteins at certain lysine residues [1]. Methylation can be carried out by two mechanisms; maintenance and de novo methylation. The former replicates methylation patterns after DNA replication on the daughter strand, copied from the parental. De novo methylation concerns the addition of new methyl groups to cytosine nucleotides at CpG rich areas of the genome[1]. The results of these processes are the silencing of genes by methylation and subsequent deacetylation of histone lysine residues. The reverse is also true for the demethylation and acetylation of histones, resulting in gene activation. Patterns of methylation have been observed frequently in cancer cell lines, leading to the hypothesis that de novo hypo- and hypermethylation may be a causal mechanism in tumourigenesis[2].
Mechanisms of epigenetic modification
Methylation of CpG dinucleotides is catalysed by DNA methyltransferases (Dnmt’s), whilst histone acetylation is catalysed by histone acetyltransferases (HAT’s) [3] (fig1). Lysine deacetylation is carried out by histone deacetylases (HDAC’s), which is linked with chromatin condensation and formation of heterochromatin [3]. This mechanism of deacetylation may have primarily evolved as a mechanism to prevent over expression of genes in the wake of transcription elongation by DNA polymerases [4].
Additionally it has been observed that histone lysine residues can also undergo methylation by histone methyltransferases (HMT’s), resulting in either activation or inhibition of transcription. To add to this further complexity lysine residues can be mono-, di- or tri-methlyated, which has been thought to be involved in longer term epigenetic “memory” [5].
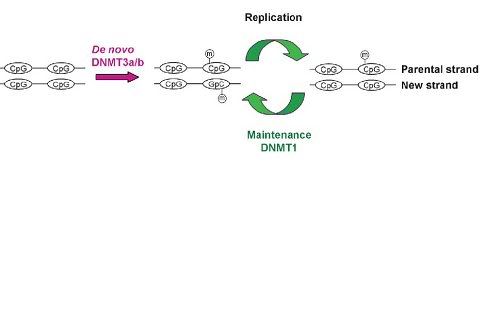
Fig1. Mechanisms for de novo and maintenance methylation byDNMT3a/b and Dnmt1 respectively during DNA replication prior to cell division [1].
Cancer and epigenetics
The first epigenetic modifications in cancer cells were observed more than two decades ago by Feinberg and Vogelstein [6]. There are two proposed mechanisms involved in cancer; hypomethylation in repetitive sequences resulting in chromosomal instability, and hypermethylation of CpG dinucleotides in promoter sequences of tumour suppressor genes [7, 8]. Hypermethylation of tumour suppressor genes has been observed resulting in silencing of transcription and subsequent deacetylation of histone lysine residues. The areas where epigenetic modification has been observed primarily concern cell checkpoint pathways, cell adhesion and invasion, activation and regulation of apoptosis, DNA repair pathways and response to growth factors [9]. Studies carried out to elucidate gene expression patterns by epigenetic means have identified particular mechanisms that may be universal to breast cancer cell lines. Munot et al show that epigenetic silencing of O6-methylguanine-DNA methyltransferase (MGMT) is linked to high grade breast tumour, and may well be a target of future therapy [1, 10].
The Knudson two-hit hypothesis proposes that cancer requires the loss of both alleles of a tumour suppressor gene to initiate tumourigenesis [11] (fig2). Although the first of these requires a heritable or somatic mutation, the second “hit” can be induced by hypermethylation and deacetylation.
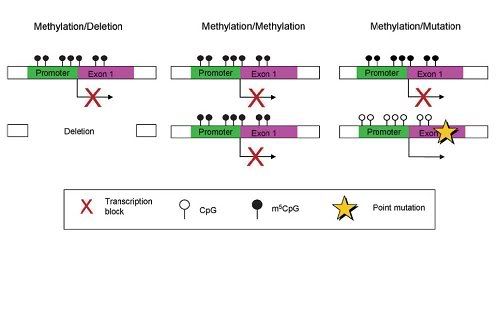
Fig2. Potential mechanisms for the Knudson two hit hypothesis involving promoter hypermethylation as a way of inactivating one allele. Mutation and hypermethylation can occur in either order [1].
To test the hypothesis of global hypomethylation, mouse models were constructed which displayed affected cancer incidence, confirming that hypomethylation leads to genomic instability [12]. Additionally polymorphisms within the gene for the enzyme responsible for de novo methylation, Dnmt3b, have been identified as a potential marker of lung cancer susceptibility [13].
Promoter sequence methylation has been linked to the silencing of tumour suppressor genes [1, 8]. Hypermethylation of CpG islands is primarily observed where heritable susceptibility is a causal mechanism of tumourigenesis. Many of the best studied tumour suppressors that undergo mutational inactivation have been observed to be silenced by promoter hypermethylation, including BRCA1 and p16INK4A [1, 8].
It has been hypothesised that hypermethylation of promoters during early stem cell differentiation may confer a selective survival or growth advantages [14]. In a subset of colorectal cancers, systematic inactivation of tumour suppressors by hypermethylation has been observed, even within the same tumour [15]. This observation has been designated the ‘CpG island methylator phenotype’ (CIMP). Most notably this phenotype has been associated with particular mutations of the oncogene BRAF, and microsatellite instability from methylation of the promoter sequence of the mismatch repair pathway gene MLH1. CIMP has also been recorded in other cancer types, including; acute myeloid leukaemia (AML), acute lymphoblastic leukaemia (ALL), oesophageal, glioblastomas, gastric, liver, pancreatic and ovarian cancers (table1) [16].
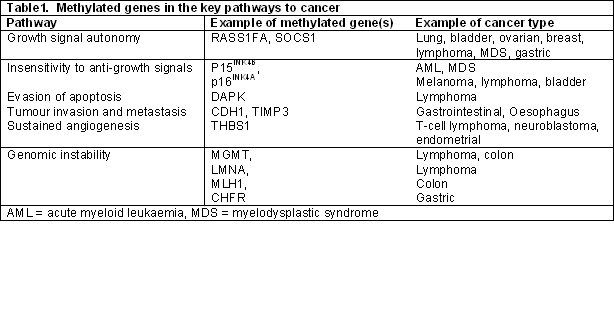
Table1. Examples of genes involved in key regulatory pathways that exhibit hypermethylation of promoter sequence CpG’s as found in varying cancer types [1].
More recently studies have shown that epigenetic silencing of microRNA’s (miRNA’s) can be observed in certain cancer types [1, 8]. The first example of this was the silencing of miR-127, a translation inhibitor of the oncogene BCL-6, which is linked to transcriptional inhibition of p53 and downstream effectors of p53 in B-cell lymphomas [17]. Reactivation of this miRNA was shown to down regulate the levels of BCL-6 without affecting the levels of mRNA. However this does not appear typical to all cancers, as Diederichs and Haber discovered inhibitory epigenetic therapy had no affect on miRNA expression in lung cancer, which indicates that different regulatory mechanism may be responsible for miRNA expression patterns depending on the malignancy type [17]. Antisense RNA silencing of p15 has recently been observed by epigenetic means. Yu et al observed that antisense RNA can lead to transcriptional inhibition of its sense tumour suppressor gene, opening up another mechanism by which RNAi pathways may be linked to the induction of epigenetic modification of tumour suppressors [18].
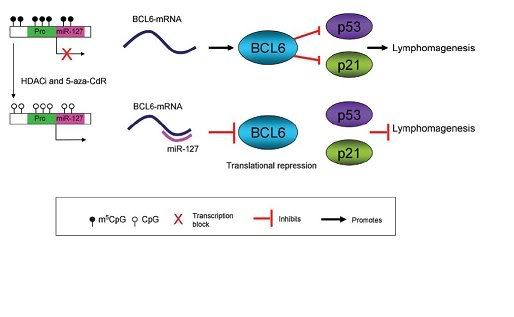
Fig3. Translational inhibition of mRNA by RNAi involving miR-127, and inactivation of micro-RNA transcription by promoter methylation. BCL6 has been shown to down regulate p53 and its downstream effector p21, leading to lymphomagenesis. The chemotherapeutic drug 5-aza-CdR can lead to reactivation of miR-127 by inducing expression of the gene [1].
The future of epigenetics and cancer
Epigenetics has become one of the most important emerging fields in the biosciences. It has opened up a window into further evolutionary mechanisms of gene expression regulation, as well as methylation and deacetylation enzymes becoming ideal targets for cancer therapy and as diagnostic tools.
Many recent studies have shown that epigenetic mechanisms are displayed and have been observed in differing malignancy types, leading to the confirmation that these have casual mechanisms in the progression of cancer. They may in fact be linked to several regulatory processes including the cells native RNAi pathways, as well as vital to the survival prospects of cancerous cells. Although so far studies have not revealed the extent to which hypermethylation leads to cancer progression, invasion and metastasis.
In the future we may well see the first ‘methylomes’ published as companions to the genome, and the information contained within may well lead to customised therapies for more than just cancer.
Abbreviations
ALL – acute lymphoblastic leukaemia
AML – acute myeloid leukaemia
CIMP – CpG-island methylator phenotype
DNMT – DNA methyltransferase
HAT – Histone acetyltransferase
HDAC – Histone deactylase complex
HMT – Histone methyltransferases
m5CpG – 5-methlyl CpG
MGMT – O6-methylguanine methyltransferase
miRNA – micro RNA
RNAi – RNA interference
References
[1] Grønbæk. K., Hother. C., Jones. P.A., (2007), Epigenetic changes in cancer, APMIS 115, 1039-1059
[2] Hanahan. D. and Weinberg. R.A, (2000), The hallmarks of cancer, Cell 100, 57-70
[3] Klose. R.J. and Bird .A.P, (2006), Genomic DNA methylation; the mark and its mediators, TRENDS 31 (2), 89-97
[4] Mellor.J., (2006), Dynamic nucleosomes and gene transcription Trends Genet 22 (6), 320-329
[5] Jenuwein. T., (2006), The epigenetic magic of histone lysine methylation FEBS J 273 (14), 3121-3135
[6] Feinberg. A.P. and Vogelstein. B., (1983) Hypomethylation distinguishes gene of some cancers from their normal counterparts, Nature 301 (5895), 89-92
[7] Esteller. M., (2005) Aberrant DNA methylation as a cancer-inducing mechanism, Annu Rev Pharmacol Toxicol 45, 629-656
[8] Lohrum. M., Stunnenberg. H.G., Logie. C., (2007) The new frontier in cancer research: Deciphering cancer epigenetics, IJBCB 39, 1450-1461
[9] Keshet. I, Schlesinger. Y., Farkash. S., Rand. E., Hecht. M., Segal. E. et al (2006) Evidence for an instructive mechanism of de novo methylation in cancer cells, Nat Genet 38 (2), 149-153
[10] Munot. K., Bell. S.M., Lane. S., Horgan. K., Hanby. A.M., Speirs. V., (2006) Pattern of expression of genes linked to epigenetic silencing in human breast cancer, Human Path 37, 989-999
[11] Knudson. A.G., (2001) Two genetic hits (more or less) to cancer, Nat Rev Cancer 1(2), 157-162
[12] Gaudet. F., Hodgson. J.G., Eden. A., Jackson-Grusby. L., Dausman. J., Gray. J.W., et al, (2003) Induction of tumours in mice by genomic hypomethylation, Science 300 (5618), 489-492
[13] Roberts. C.W., & Orkin. S.H., (2004) The SWI/SNF complex – Chromatin and cancer, Nat Rev Cancer 4 (2), 133-142
[14] Baylin. S.B., & Ohm. J.E., (2006) Epigenetic silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 6, 107-116
[15] Toyota. M., Ahuja. N., Ohe-Toyota. M., Herman .J.G., Baylin S.B., Issa. J.P., (1999) CpG island methylator phenotype in colorectal cancer, Proc Natl Acad Sci USA 96, 8681-8686
[16] Issa. J.P., CpG island methylator phenotype in cancer, Nat Rev Cancer 4, 988-993
[17] Diederichs. S., Haber. D.A., (2006) Sequence variations of microRNA’s in human cancer: alterations in predicted secondary structure do not affect processing, Cancer Res 66, 6097-6104
[18] Yu. W., Gius. D., Onyango. P., Muldoon-Jacobs. K., Karp. J., Feinberg. A.P., Cui. H., (2008) Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA, Nature 451 (10), 202-207





